
1 Trim adapters and remove low sequencing quality using Cutadapt+Trimmomatic OR trim_galore (example).
cutadapt -a CTGTAGGCACCATCA --error-rate=0.1 --overlap=3 --times=2 --output=sample.trimed.fastq.gz sample.fastq.gz
--error-rate: Maximum allowed error rate as value between 0 and 1 (no. of errors divided by length of matching region) (0.1 = 10%)
--overlap: MINLENGTH Require MINLENGTH overlap between read and adapter for an adapter to be found.
--times: Remove up to COUNT adapters from each read or number of round for adapter finding and removal
java -jar /path/trimmomatic-0.39.jar SE -phred33 sample.trimed.fastq.gz sample.trimed.cleaned.fastq.gz LEADING:30 TRAILING:30 MINLEN:18
-phred33: Sequence quality type which can be infer using FastQC
LEADING: Cut bases off the start of a read, if below a threshold quality
TRAILING: Cut bases off the end of a read, if below a threshold quality
MINLEN: Drop the read if it is below a specified length
-
trim_galore sample.trimed.fastq.gz --phred33 CTGTAGGCACCATCA --length 18 -e 0.1 -q 30 --stringency 3 -o outdir
--phred33: Sequence quality type which can be infer using FastQC
--length: Discard reads that became shorter than length INT because of either quality or adapter trimming. A value of '0' effectively disables this behaviour.
-e: same as --error-rate in cutadapt
-q: Phred score cut-off
--stringency: same as --overlap in cutadapt
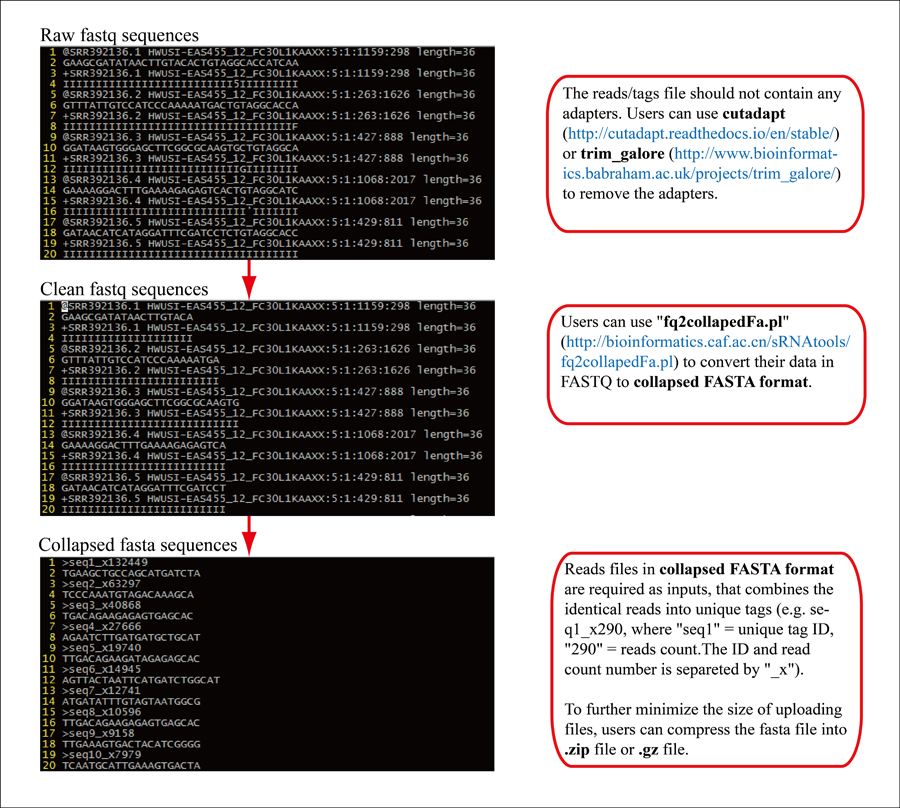
2 Generate collapsed FASTA file.
-i: input cleaned FASTQ sequences (sample_trimmed.fq.gz for trim_galore output)
-o: output collapsed FASTA file
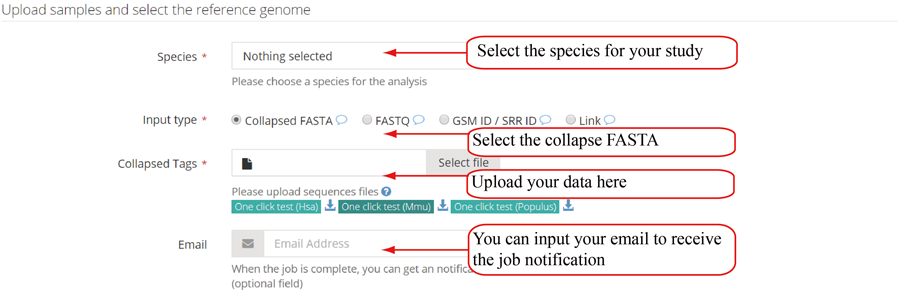
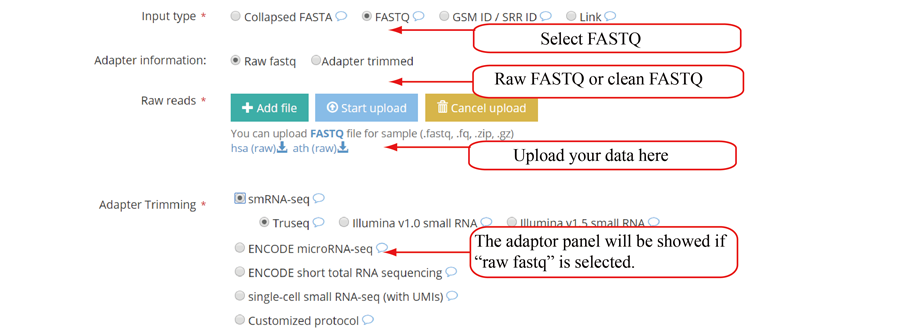

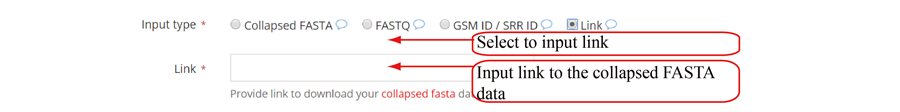

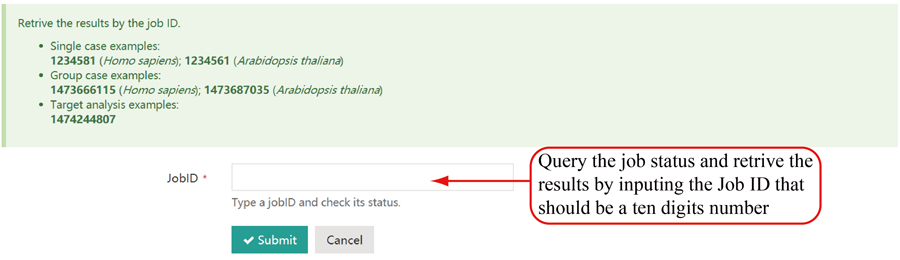
Query the job status and retrieve the results
Query the job status
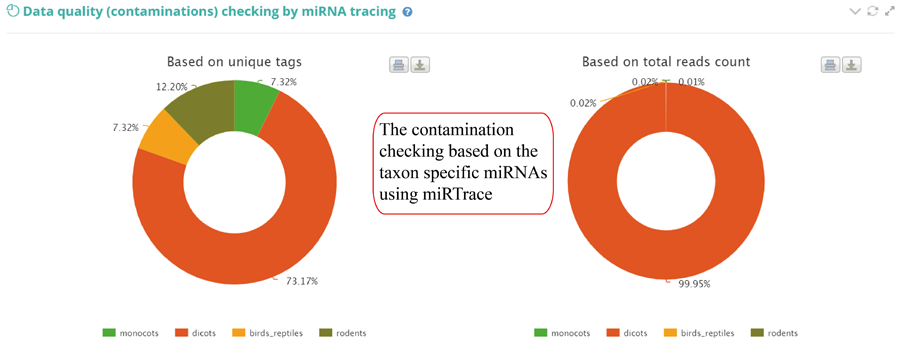
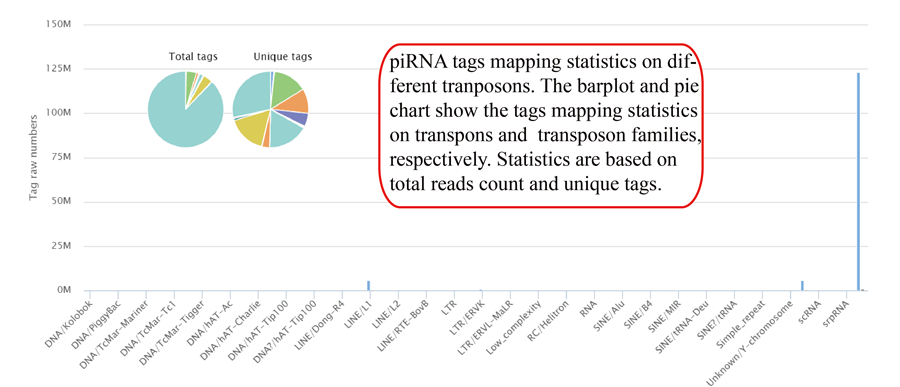
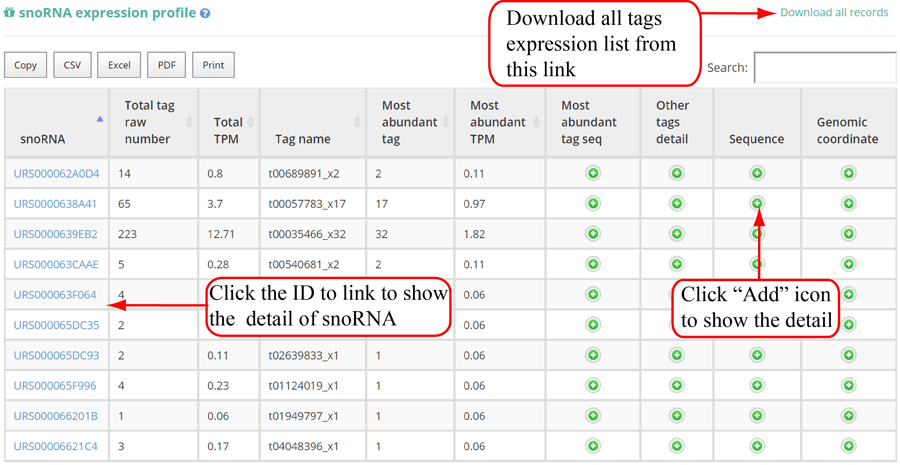
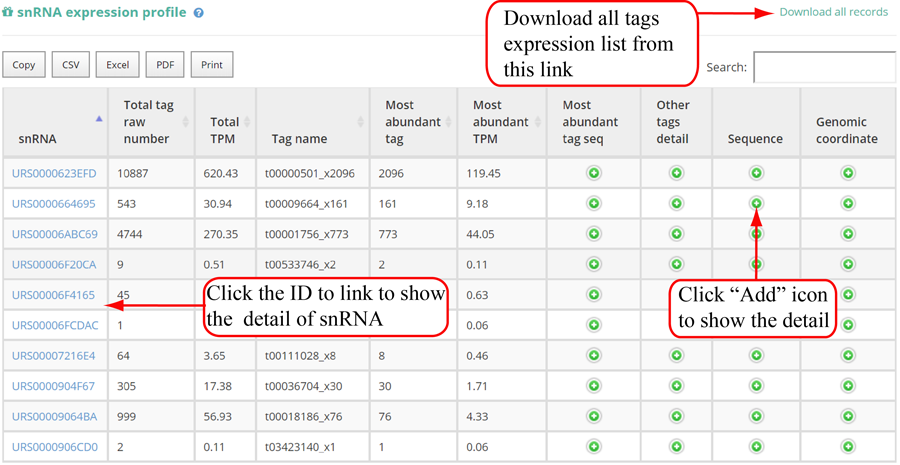
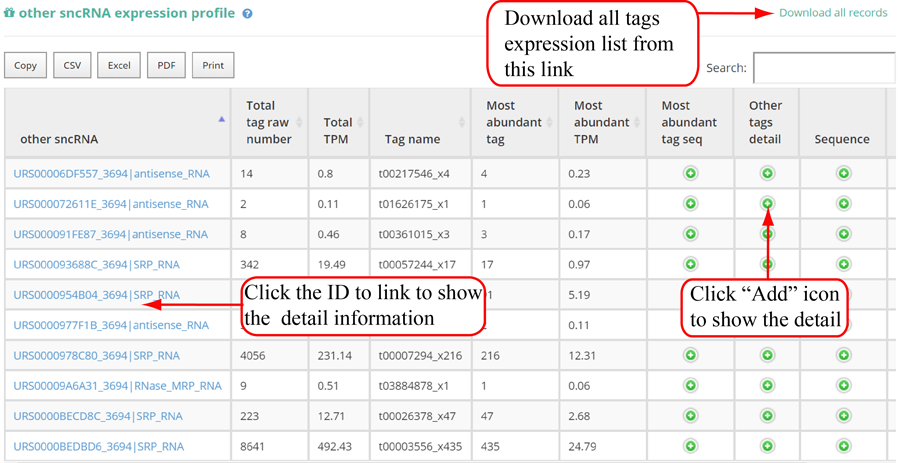
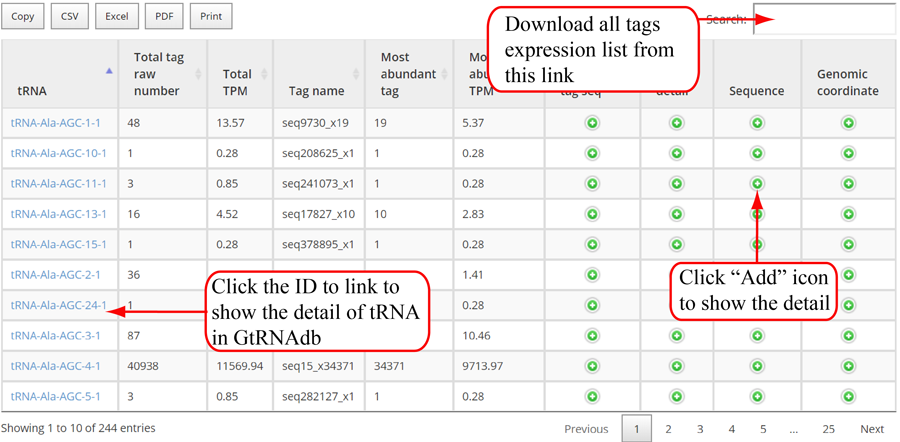
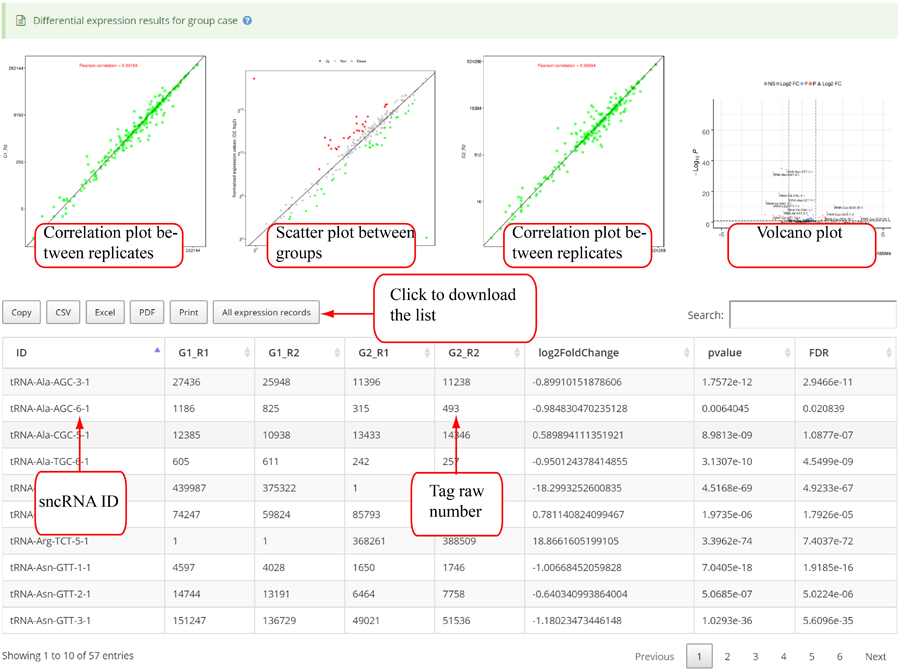
Tags mapping statistics on different types of RNAs, including miRNA, tRNA, rRNA, snRNA, snoRNA, other RFAM ncRNA, mRNA, lncRNA, other mapping.

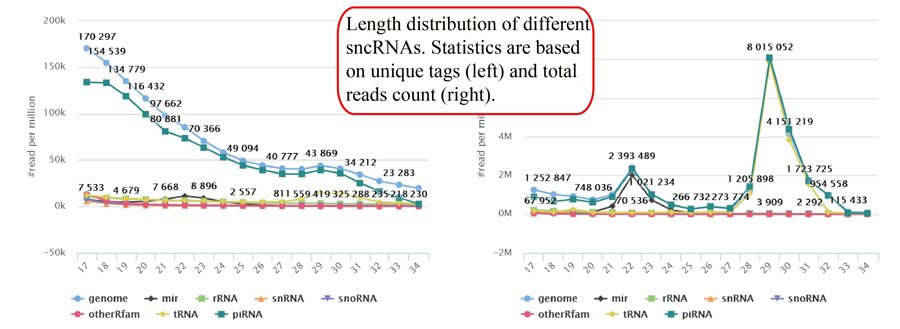
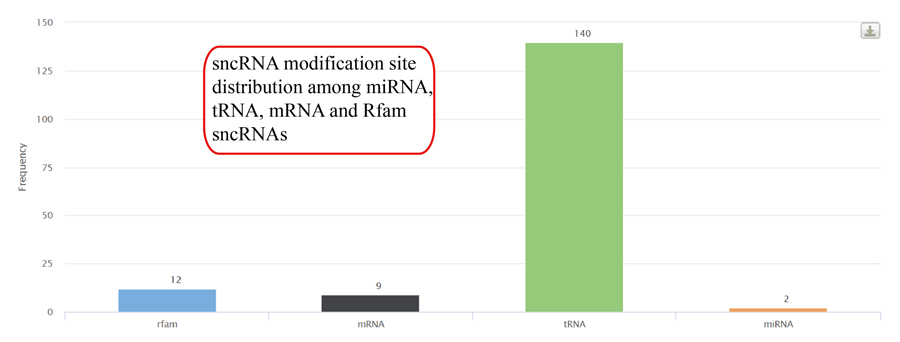
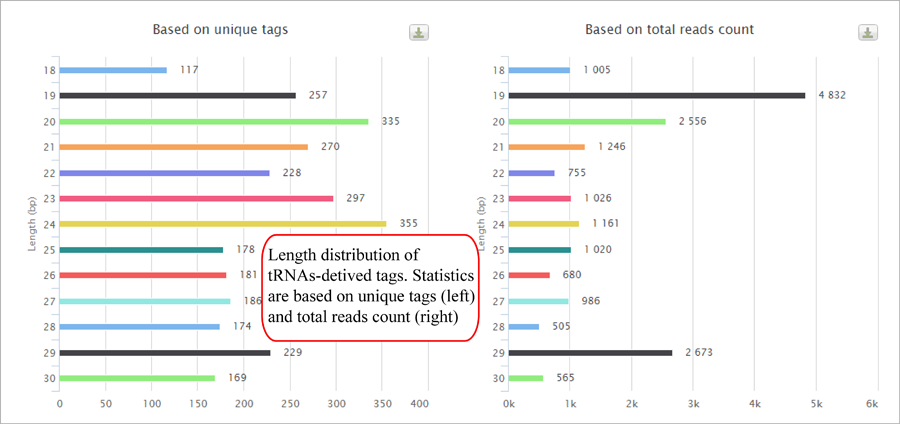
Tag length distribution of different sncRNAs. Statistics are based on unique tags and total tag counts.
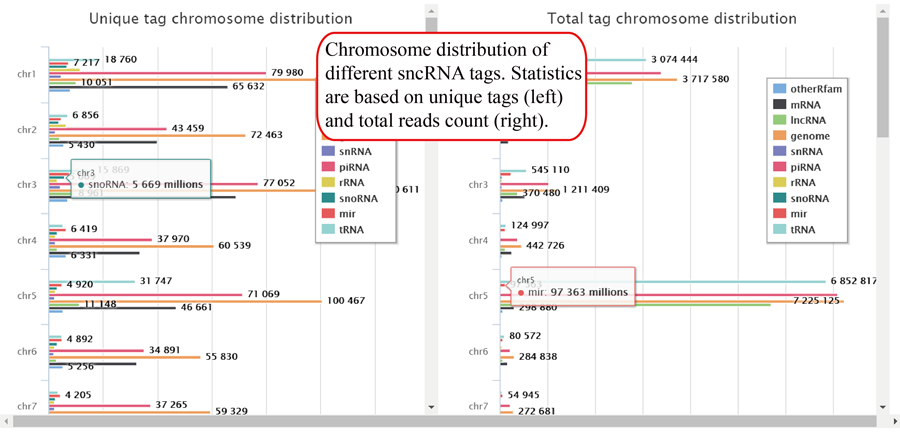
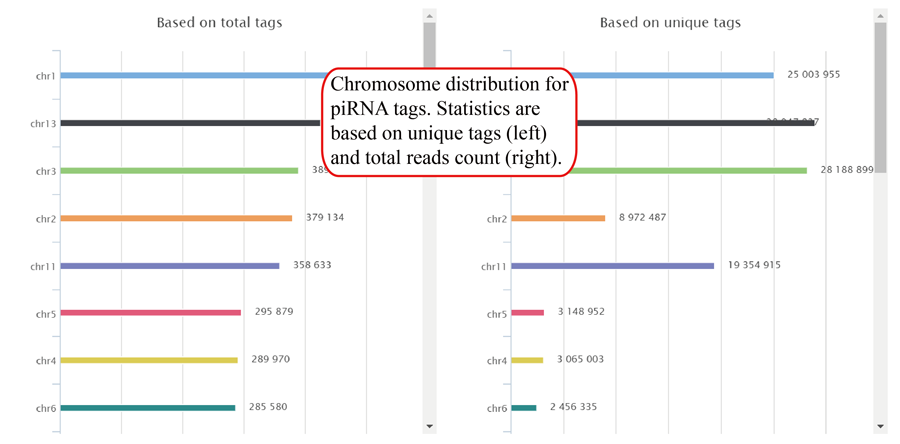
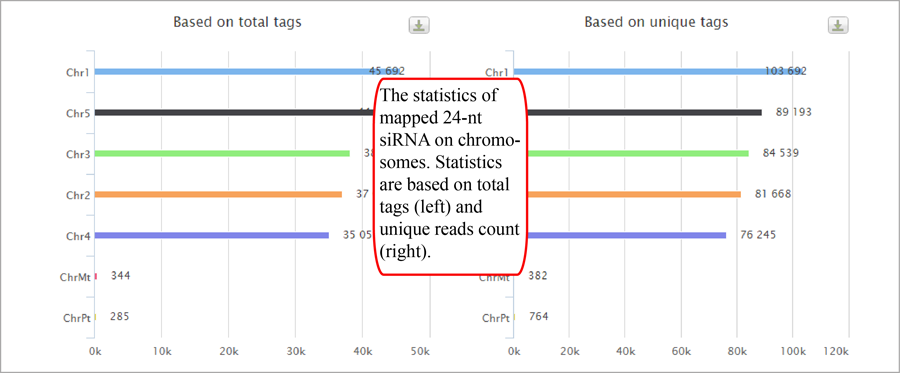
Tag length distribution of different chromosomes. Statistics are based on unique tags and total tag counts.
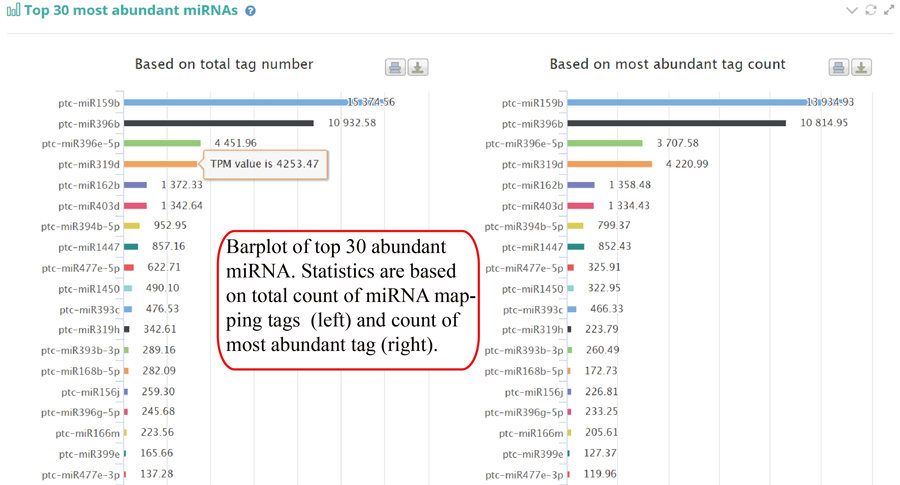
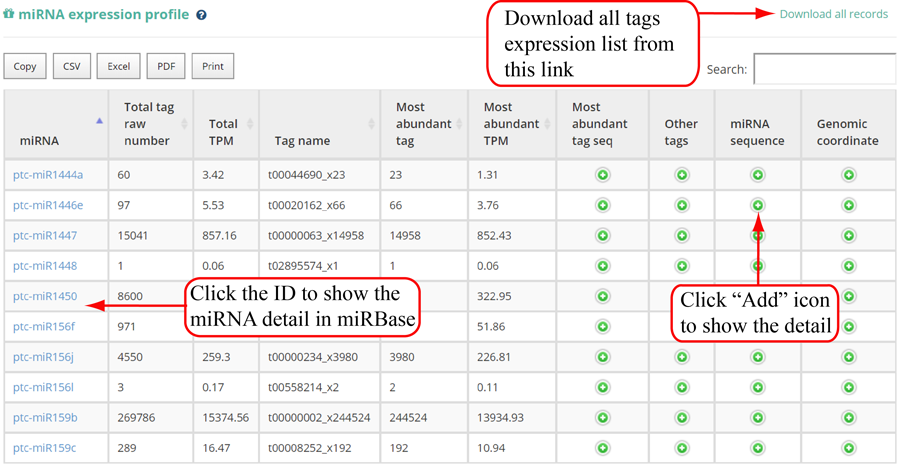
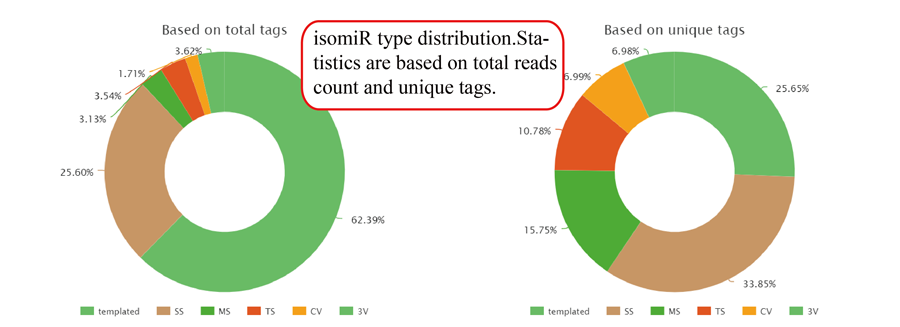
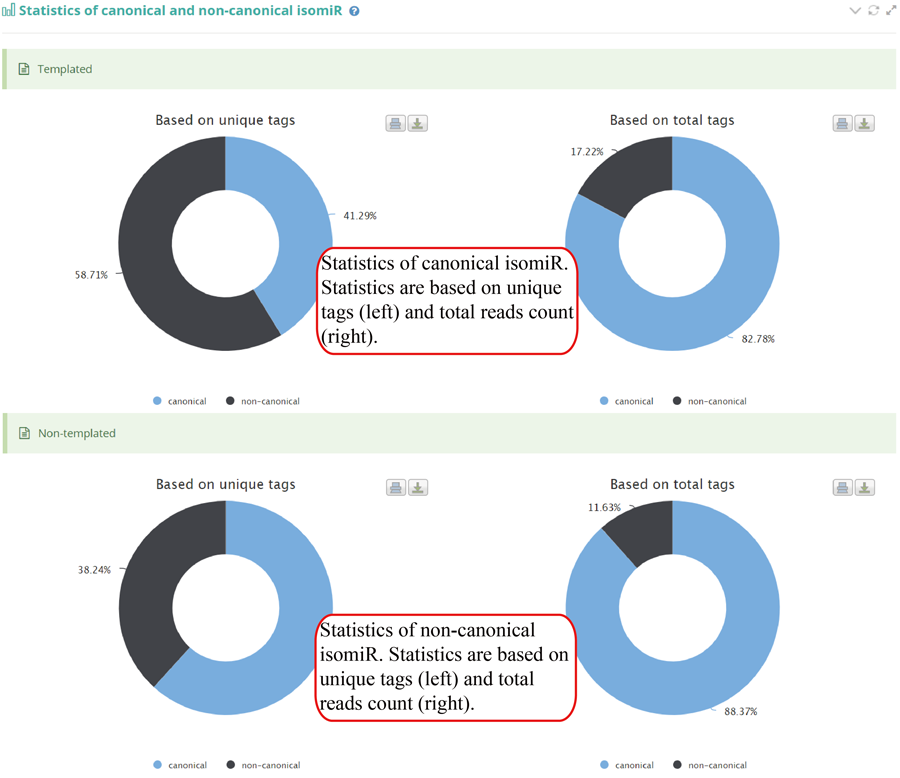
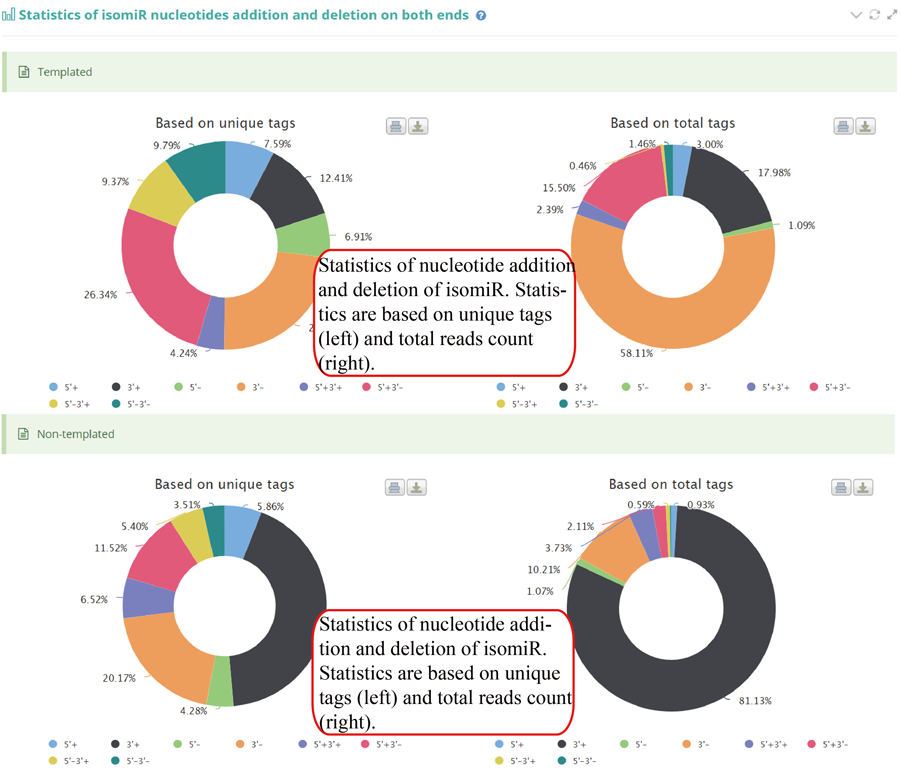
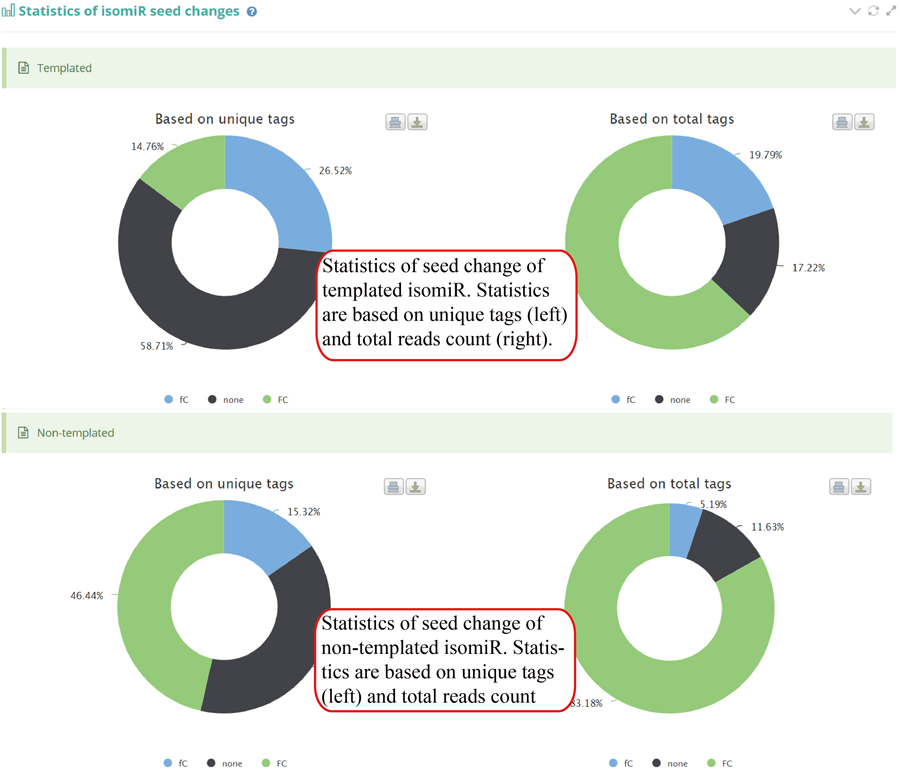
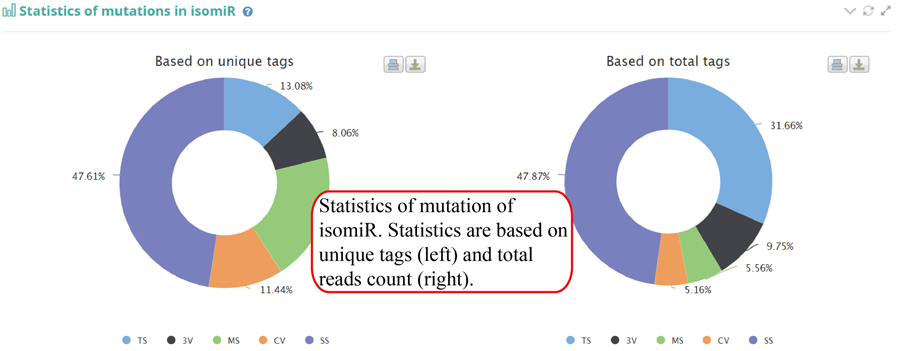
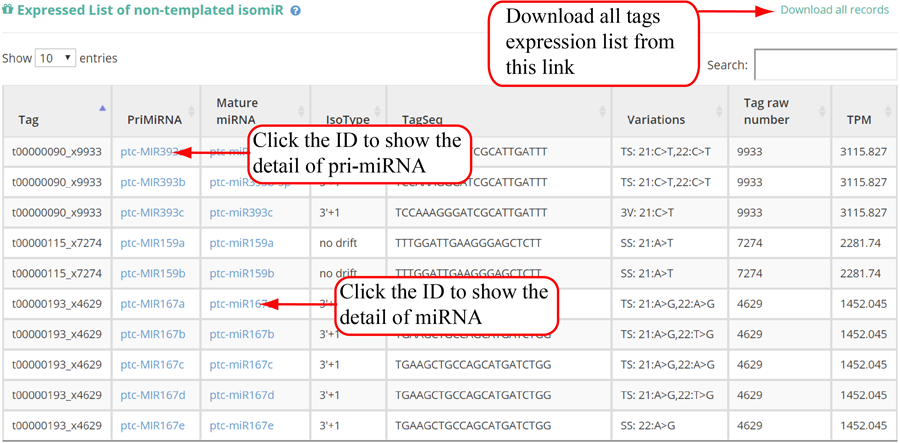
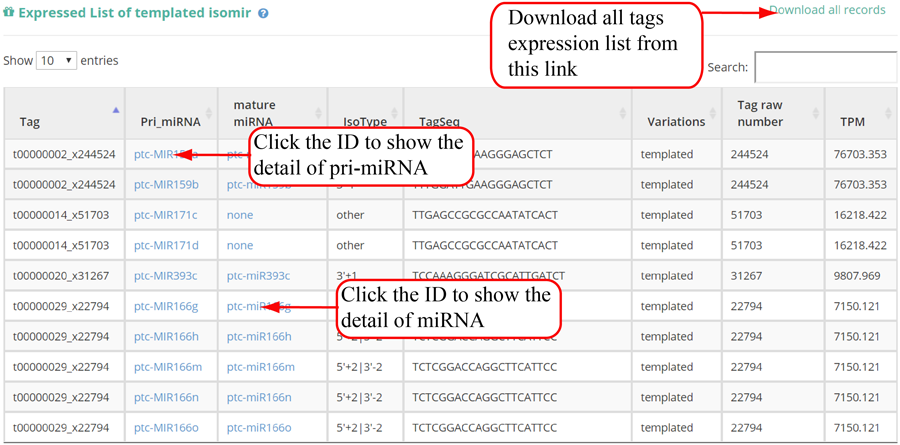
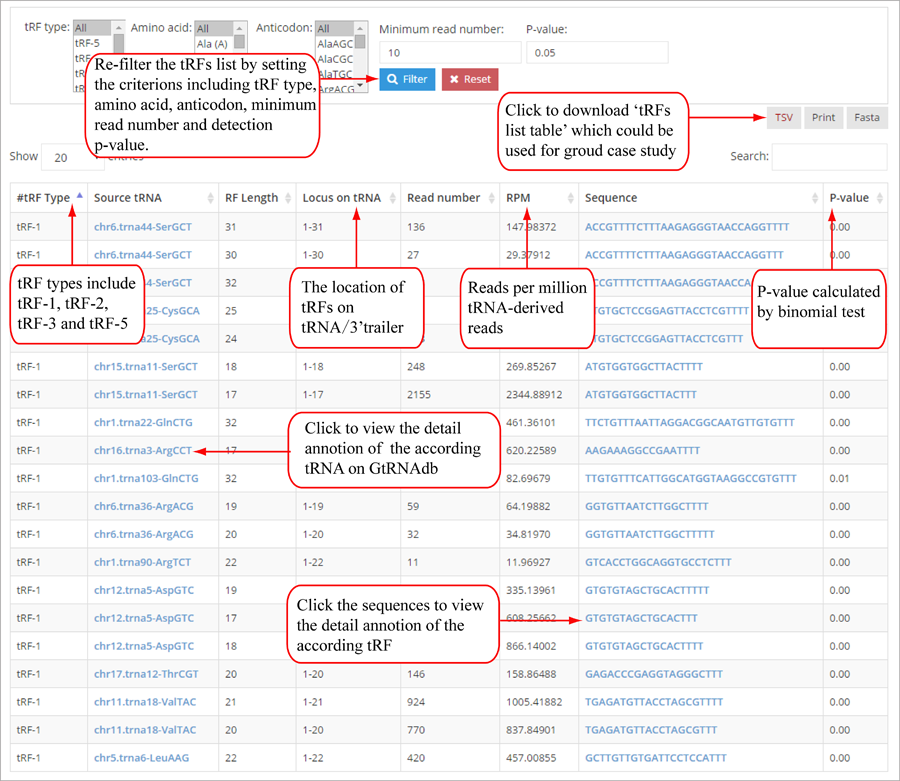
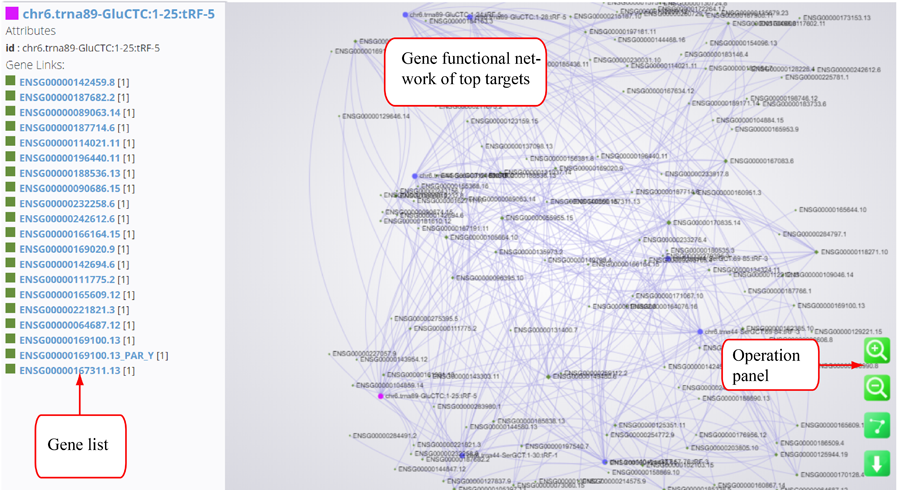
Tag distribution of isomiR, including templated isomiR and non-templated isomiR (SS, MS, TS, CV and 3V)
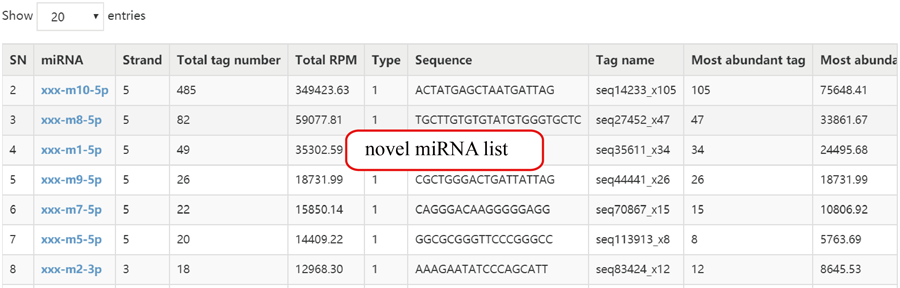
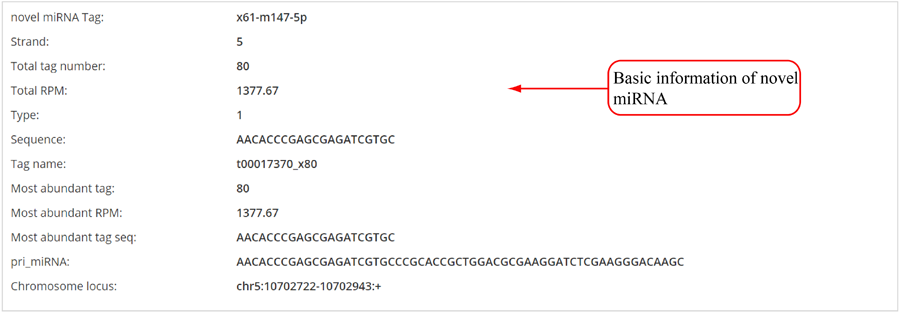
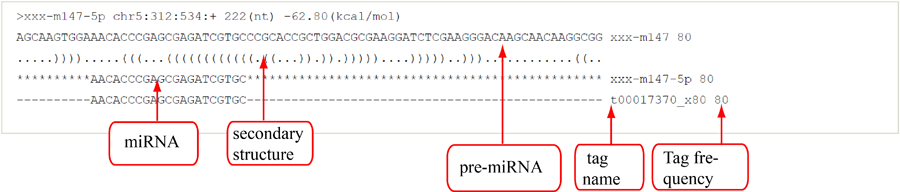
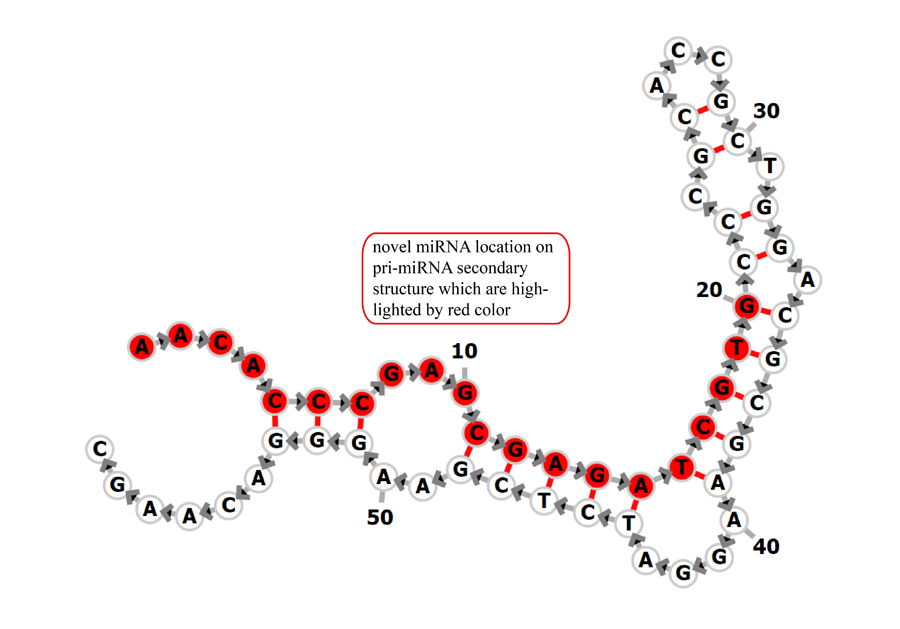
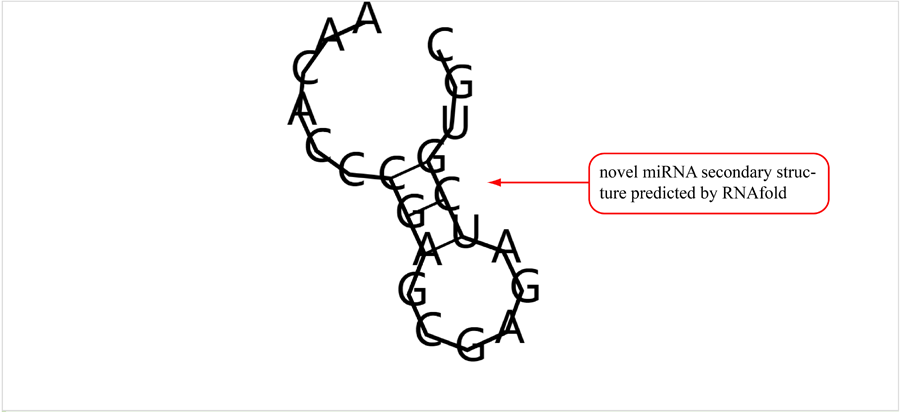
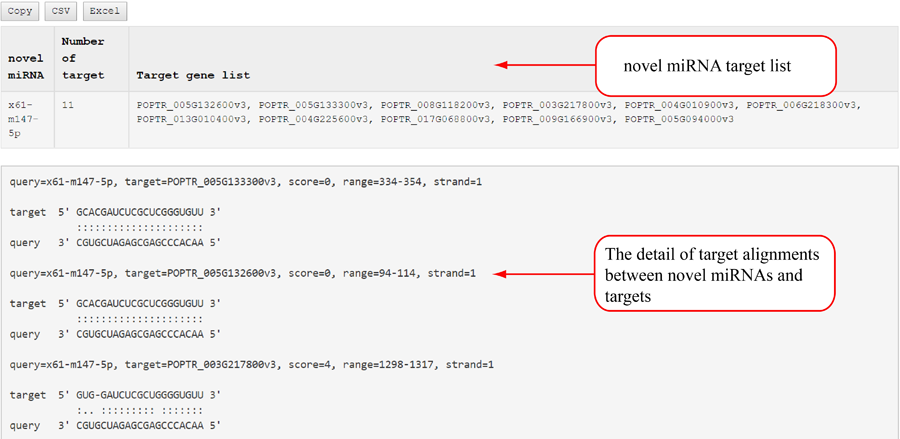
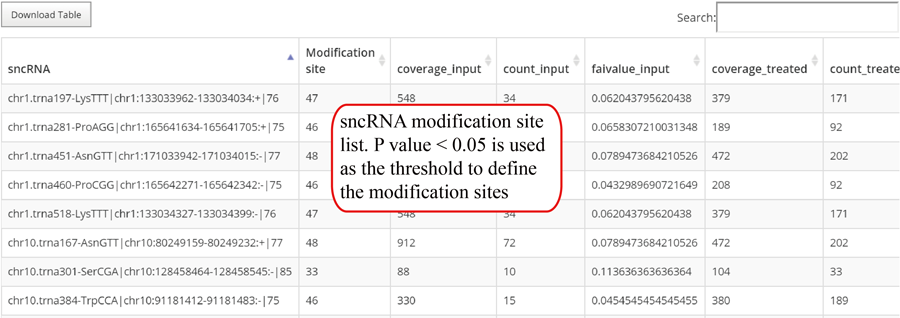
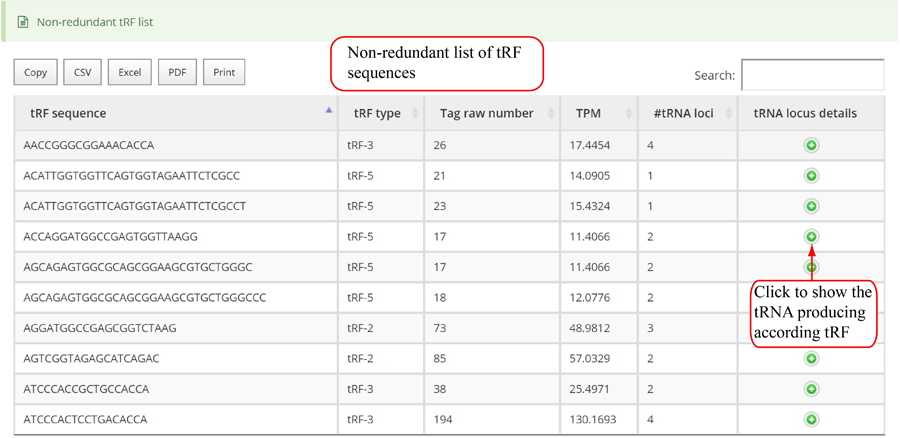
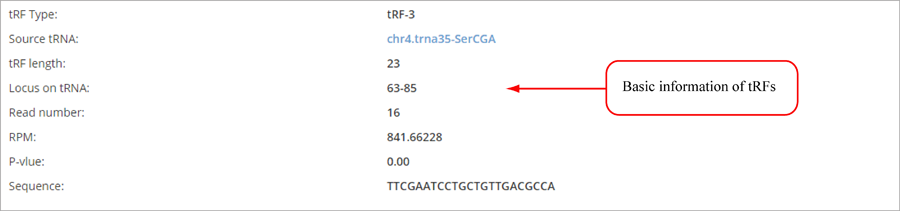
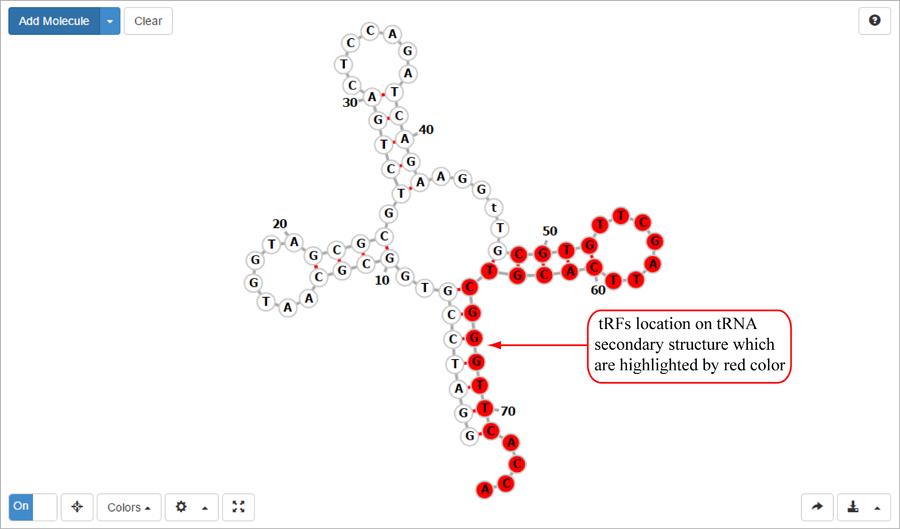
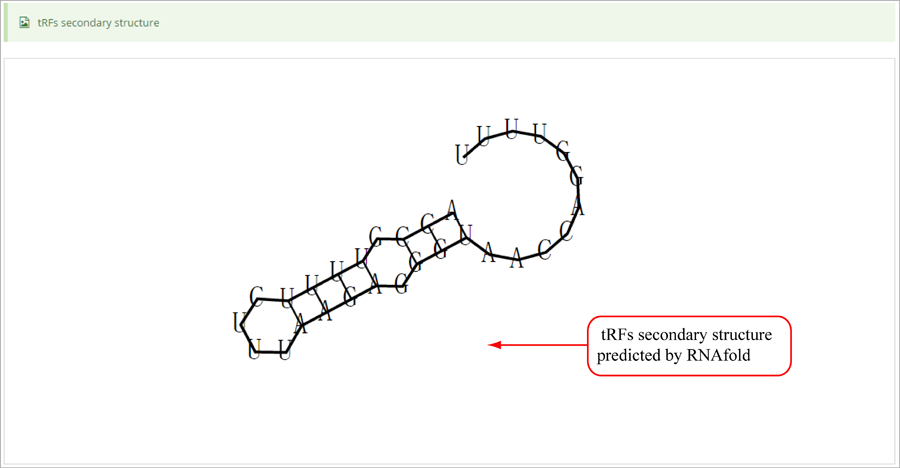
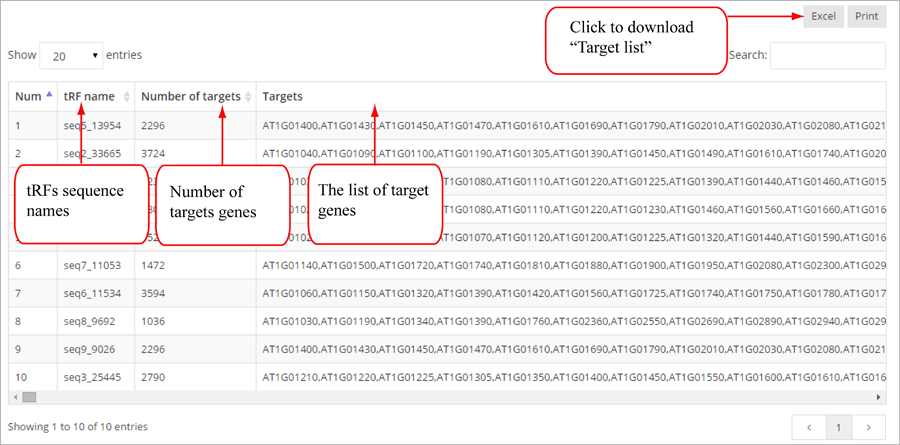
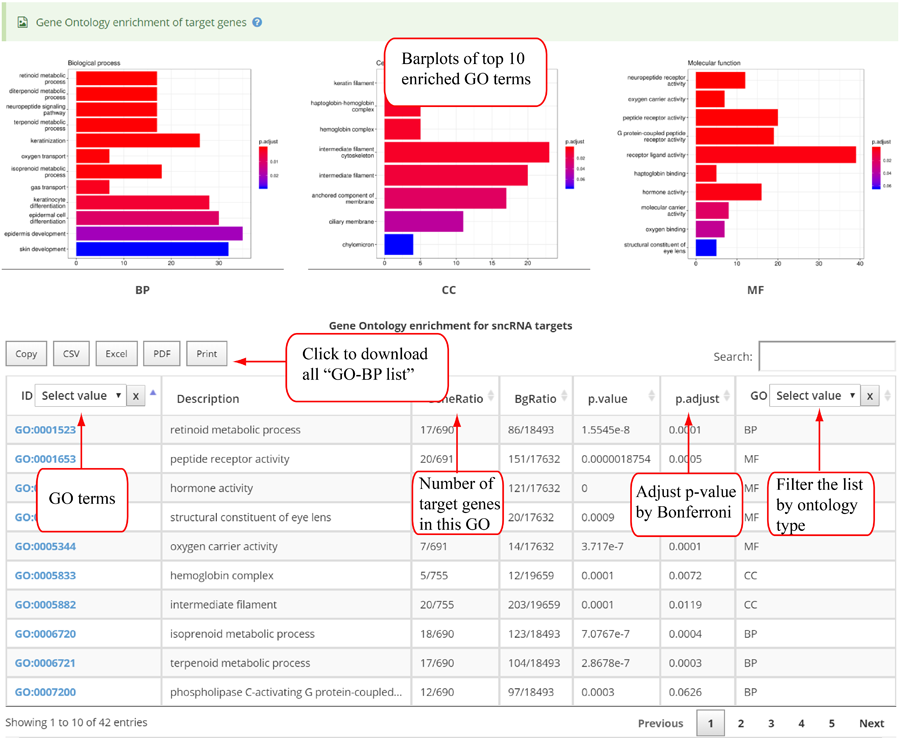
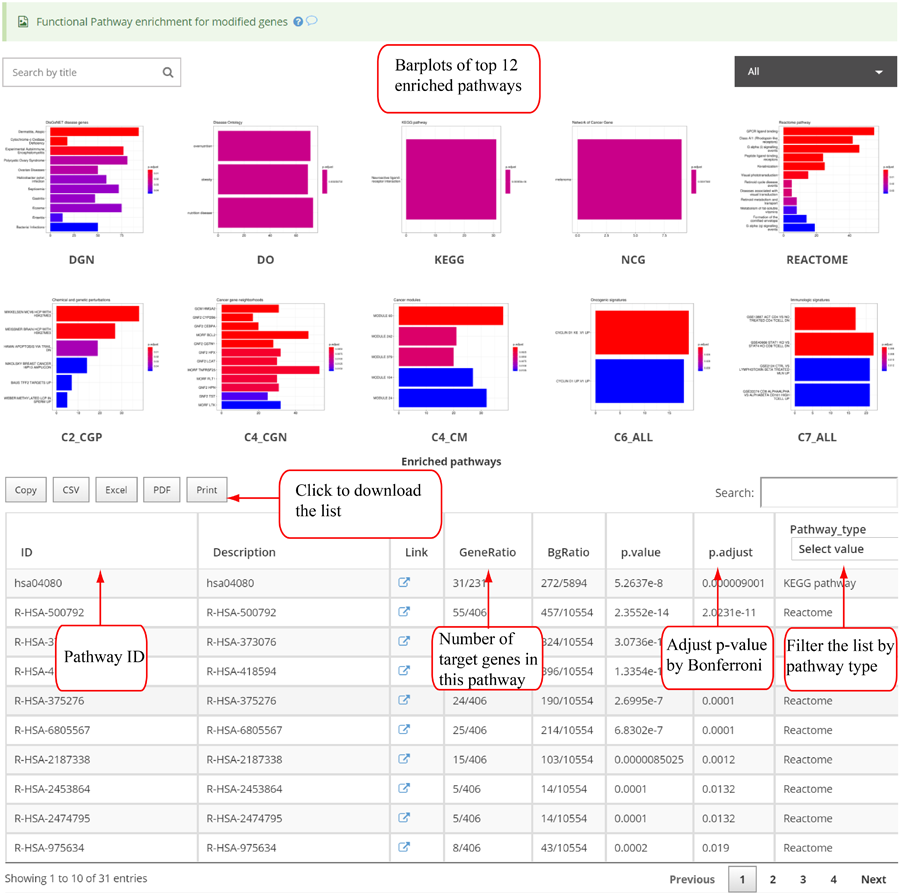
Novel miRNA list. All unclassified reads were considered for detecting candidate novel miRNA genes. Sequence of predicted putative miRNA and miRNA star along with the corresponding tag number, tag count and hairpin structure are provided.





| Browser name | Version |
|---|---|
| Internet Explorer | 11 |
| Chrome | 70.0.3538.80 |
| Firefox | 66.0 |
| Safari | 5.1.7 |